|
|
| 当前位置: 首页>首页>消费提示>医疗器械 |
| 医疗器械警戒快讯 2022年第6期(总第184期) | |||
| 分享到: | |||
| 发布日期:2022-06-24 | |||
|
医疗器械警戒快讯 2022年第6期 (总第184期) 内容提要 l 美国FDA发布关于Abbott Medical公司因标识条松动可能导致患者伤害的风险召回Dragonfly OpStar成像导管的警示信息 l美国FDA发布关于ArjoHuntleigh Polska公司因电池耗尽冒烟或起火的风险召回Sara Plus地面升降机的警示信息 l澳大利亚TGA发布关于Medtronic公司召回胰岛素泵的警示信息 l美国FDA发布关于Illumina公司测序仪具有网络安全漏洞相关风险的警示信息 l美国FDA发布关于Philips Respironics公司因电源问题意外停机的风险召回V60和V60 Plus呼吸机的警示信息 l美国FDA发布关于Atrium Medical公司因球囊或导管毂分离导致患者受伤的风险召回iCast覆膜支架的警示信息 l美国FDA发布关于Medtronic公司因泵焊接缺陷召回HVAD泵植入组件的警示信息 国家药品不良反应监测中心 国家药品监督管理局药品评价中心 http://www.cdr-adr.org.cn 美国FDA发布关于Abbott Medical公司因标识条松动可能导致患者伤害的风险召回Dragonfly OpStar成像导管的警示信息 发布日期:2022年5月26日 召回级别:I级,是最严重的召回类型,使用这些器械可能造成严重伤害或者死亡。 产品信息: l产品名称:Dragonfly OpStar成像导管 l产品批号详见网站: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=193136 l在美国召回的数量:4800 l召回发起日期:2022年4月11日
产品用途:带有光学相干断层成像术(OCT)成像系统的Dragonfly OpStar成像导管用于为适合进行基于导管的微创介入手术的人提供向心脏(冠状动脉)输送血液和氧气的血管成像,以治疗冠状动脉疾病。 召回原因:因为距离导管尖端最远的标记条(近端标记)可能会变松,Abbott Medical公司正在召回某些批次的Dragonfly OpStar成像导管,目前已经在2个案例中观察到标记条与导管分离。 移除导管后,与设备分离的标记条可能会留在体内,导致血管损伤,包括但不限于栓塞(血管阻塞)、血栓形成(血凝块)、夹层(撕裂)、缺血(心脏供血不足)、梗塞(心脏病发作)、感染或者死亡。 目前,已有5起事件和1例伤害报告与此设备问题相关,尚无与使用该设备相关的死亡事件。 受影响人群: l使用Dragonfly OpStar成像导管进行冠状动脉OCT成像的医务人员。 l将使用Dragonfly OpStar成像导管对冠状动脉进行基于导管的OCT成像的患者。这个问题不会影响已经做过这个手术的人。 采取措施:2022年4月11日,Abbott Medical公司向所有收到受影响设备的客户发送了一份紧急医疗器械召回通知,包含以下说明内容: l立即停止使用受影响批次的设备。 l查看库存并填写紧急医疗器械召回通知中包含的有效性检查表。 l将所有未使用的受影响设备退还给Abbott Medical公司。 l与所有相关人员共享此信息。 l通知任何可能通过其他分销或者转让收到这些受影响产品的人。 l向Abbott Medical公司报告任何产品性能问题或者不良事件。 (美国FDA网站) 美国FDA发布关于ArjoHuntleigh Polska公司因电池耗尽冒烟或起火的风险召回Sara Plus地面升降机的警示信息 发布日期:2022年5月27日 召回级别:I级,是最严重的召回类型,使用这些器械可能造成严重伤害或者死亡。 产品信息: l产品名称:Sara Plus地面升降机 l产品序列号详见网站: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=192990。 l生产日期:2016年10月13日至2022年1月13日,2022年3月4日前收到的备件(PCB)。 l在美国召回的数量:1929(包括受影响的备件)。 l召回发起日期:2022年4月8日 产品用途:ArjoHuntleigh Polska公司Sara Plus主动式地面升降机用于患者的短时间转移,例如将患者从床上抬到轮椅或者从轮椅到卫生间。它适用于医院、疗养院和其他医疗机构。
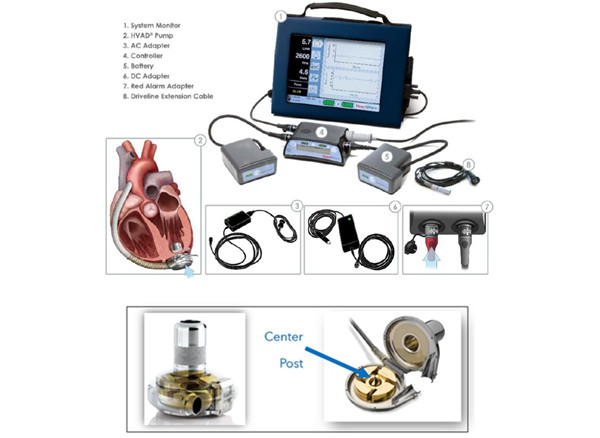
召回原因:由于此前曾多次收到该产品发生冒烟或者起火的投诉事件,ArjoHuntleigh Polska公司正在召回Sara Plus主动式地面升降机。当电池电量不足时,设备的电路板可能会过热,从而导致冒烟和/或起火。如果发生这种情况,使用该升降机或者靠近的任何人都可能受伤,包括吸入烟雾和/或烧伤。 目前已收到44起有关此问题的投诉,尚无与使用此设备有关的伤害或者死亡报告。 受影响人群: l协助患者使用Sara Plus地面升降机的医务人员。 l使用受影响的Sara Plus地面升降机的人员。 采取措施:2022年4月8日,ArjoHuntleigh Polska公司向所有收到受影响设备的客户发出紧急医疗设备召回通知,并附上可能受影响的设备清单,为所有客户和医务人员/设备用户提供以下指导。 给所有客户的建议 l确保Sara Plus地面升降机的所有护理人员和/或用户都知晓该问题。 l通过升降机上可充电电池组后面的序列号或者根据设备显示屏的显示信息来识别受影响的设备。 l为ArjoHuntleigh Polska服务技术人员制定一份计划,通过填写随信附上的客户回复表对该问题进行产品纠正,并希望Arjo联系工厂安排免费纠正。 给医务人员/设备用户的建议 l如果使用时出现烟雾,立即停止使用,按下紧急停止按钮。从电池插座中取出电池,以防止任何进一步的故障。 l在任何情况下都不要让病人无人看管。 l不要在潮湿的地方使用该设备或者向其盖喷洒任何液体。意外进水可能导致电路板故障。 l避免超载。使用转运和行走吊索进行转运时,安全工作负载为140kg(308Ibs);当使用相同的吊索进行步行练习时,安全工作负载为190kg(420Ibs)。 l请勿在电池电量耗尽或者损坏(超过2年)的情况下使用升降机,因为这可能会损坏电路板(电池的标签上可以看到电池的生产日期)。 l清洁时,请使用加入了温和清洁剂的温水用湿布擦拭。要对设备进行消毒,请先清洁设备,然后使用含有一种兼容消毒剂的溶液进行擦拭。有关详细指导,请参阅设备使用说明。 l如果设备未使用,请取出电池。 l使用设备前,请确保其电池处于良好的工作状态。 l当设备显示屏上的电池放电指示灯(小电池符号)下降到只剩八段中的三段时,立即为电池充电。 (美国FDA网站) 澳大利亚TGA发布关于Medtronic公司召回胰岛素泵的警示信息 发布日期:2022年6月1日 召回级别:I级 召回产品:所有MiniMed 640G,670G,770G和780G胰岛素泵 召回原因:Medtronic公司糖尿病信号探测注意到关于MiniMed 600系列泵电池盖裂纹/损坏(BA01)的抱怨信号。通过对返回产品的失效分析,发现电池盖接触点损坏/破裂/松动/丢失。如果黄金触点在用户更换电池时脱落,当用户放入新的AA电池时,由于电路仍然没有通电,“放入电池”的警报将不会自动清除。警报会在10分钟后升级为警报声,然后泵就会关闭,直到电路建立才会重新通电。 此种情况可能会导致严重的高血糖和糖尿病酮症酸中毒(DKA)。 采取措施:产品缺陷更正。Medtronic公司正在通知客户,告知Medtronic公司正在进行一种新的电池盖设计,将于2023年第一季度上市。 与此同时,Medtronic公司建议客户遵循客户信函中说明的临时措施(Medtronic公司仅面向受影响客户提供)。 (澳大利亚TGA网站) 美国FDA发布关于Illumina公司测序仪具有网络安全漏洞相关风险的警示信息 发布日期:2022年6月2日 美国食品药品监督管理局(FDA)正在向实验室工作人员和医护人员通报一个网络安全漏洞,该漏洞会影响Illumina NextSeq 550Dx、MiSeqDx、NextSeq 500、NextSeq 550、MiSeq、iSeq和MiniSeq等新一代测序仪中的软件。这些仪器为医疗器械,可以用于对一个人的DNA进行测序或检测各种遗传疾病等临床诊断,也可以只用于研究(RUO)。这些设备中的一些具有双引导模式,允许用户在临床诊断模式或RUO模式下操作它们。用于RUO的设备通常处于开发阶段,必须标明“仅供研究使用”“不适用于诊断程序”,尽管许多实验室可能在临床诊断测试中使用这些设备。 网络安全漏洞影响Local Run Manager(LRM)软件。未经授权的用户可以利用漏洞进行以下操作: l远程控制设备; l操作系统以更改设备或客户网络上的设置、配置、软件或数据;或者 l影响设备中用于临床诊断的患者测试结果,包括使仪器不提供结果或提供不正确的结果、改变结果或潜在的数据泄露。 Illumina开发了一个软件补丁来防止该漏洞被利用,并致力于为当前和未来的设备提供永久的软件修复。FDA希望实验室工作人员和医务人员注意到降低这些网络安全风险需要采取的措施。 建议 l查看Illumina于2022年5月3日发送给受影响客户的紧急安全通知或产品质量通知(针对RUO客户)。如果您没有收到Illumina的通知,但认为您应该收到,请联系techsupport@illumina.com。 l当连接到互联网时,立即下载软件补丁(Dx模式和RUO模式)并安装在每台受影响的设备上,包括Dx设备上RUO模式下每个独立运行的设备,如LRM。 l如果您没有连接到互联网,请联系techsupport@illumina.com获取安装软件补丁程序的其他方法的说明。 l如果您怀疑您的设备可能已被未经授权的用户破坏,请立即联系techsupport@illumina.com。 有关Illumina网络安全漏洞的更多信息,请参见网络安全和基础设施安全局(CISA)发布的公告,ICSA-22-153-02。 背景 2022年5月3日,Illumina向受影响的客户发送通知,指导他们检查相关设备和医疗器械,寻找潜在利用该漏洞的迹象。 Illumina已经开发了一个软件补丁来防止该漏洞被利用,并且正在积极地为当前和未来的设备提供一个永久的软件修复。 目前,FDA和Illumina尚未收到任何表明该漏洞已被利用的报告。 FDA采取的措施 FDA正在与Illumina合作,并与CISA协调,以识别、沟通和防止与该网络安全漏洞相关的不良事件。如果有新的或额外的信息,FDA将继续通知医护人员和实验室工作人员。 向FDA报告问题 FDA鼓励用户报告Illumina新一代测序仪器出现的任何不良事件或可疑不良事件。 l自愿报告可通过MedWatch,FDA安全信息和不良事件报告计划提交。 l设备制造商和使用单位必须遵守适用的医疗器械报告(MDR)法规。 l被遵守FDA使用单位报告要求的单位雇佣的医护人员应该遵守其所在单位建立的报告程序。 (美国FDA网站) 美国FDA发布关于Philips Respironics公司因电源问题意外停机的风险召回V60和V60 Plus呼吸机的警示信息 发布日期:2022年6月3日 召回级别:I级,是最严重的召回类型,使用这些器械可能造成严重伤害或者死亡。 产品信息: l产品名称:V60和V60 Plus呼吸机 l产品代码和序列号详见网站: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=192390(V60呼吸机) https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=192391(V60 Plus呼吸机) l分销日期:2009年5月1日至2021年12月22日 l美国召回数量:56671台,其中V60呼吸机49680台,V60 Plus呼吸机6991台。 l召回发起日期:2022年3月10日 产品用途:Philips Respironics公司的V60和V60 Plus呼吸机用于支持患者呼吸。在医务人员的指导下,该产品在医院或其他机构环境中使用,为可以自行呼吸的成人和儿童提供机械通气,例如呼吸衰竭、慢性呼吸功能不全或者阻塞性睡眠呼吸暂停的患者。 召回原因:由于内部电源波动可能导致呼吸机在有或者没有警报的情况下意外停机,Philips Respironics公司正在召回所有V60和V60 Plus呼吸机。电源波动导致备用报警控制器重新启动,这可能导致呼吸机在没有警告的情况下完全关闭。受影响的呼吸机可能会在有或者没有警报的情况下停止通气,导致患者长时间缺氧,可能引起严重的不良后果,甚至死亡。 截至2022年4月14日,已有4例伤害报告和1例死亡报告与使用召回的设备有关。 受影响人群: l使用受影响的V60或者V60 Plus呼吸机为患者提供护理的医务人员。 l使用受影响的V60或者V60 Plus呼吸机的患者。 采取措施:2022年3月14日,Philips Respironics公司向所有受影响的收货人发送了一份紧急医疗器械更正通知,公司建议客户继续使用受影响的Philips V60和V60 Plus呼吸机,并按照使用说明和以下指导使用: l将V60/V60 Plus呼吸机连接到护士呼叫/远程报警系统。如果呼吸机主警报系统未激活,公司强烈建议使用此策略向临床医生提供备用信号。连接到远程警报的说明可在操作手册的B-5节中找到:远程警报端口。 l在连接到呼吸机之前验证任何护士呼叫/远程警报设备的操作。 l响应所有警报。建议立即响应呼吸机发出的高优先级警报,并迅速响应低优先级警报。 l安装氧气分析仪/监视器。按照制造商的说明进行此设备的设置、警报和校准。 l提供脉搏血氧饱和度以告知临床医生患者病情的变化。 l确保始终可以立即使用其他通气方式。 l如果V60/V60 Plus呼吸机停机,断开患者的连接,然后立即使用备用设备开始通气。 l公司要求客户通过传真或者电子邮件确认收到召回通知。 2022年4月22日,Philips Respironics公司向所有受影响的收货人发布了紧急医疗器械更正通知的更新,如果前面提到的缓解措施不可用,建议客户进行风险/获益分析,以评估是否应继续使用受影响的器械。 (美国FDA网站) 美国FDA发布关于Atrium Medical公司因球囊或导管毂分离导致患者受伤的风险召回iCast覆膜支架的警示信息 发布日期:2022年6月8日 召回级别:I级,是最严重的召回类型,使用这些器械可能造成严重伤害或者死亡。 产品信息: l产品名称:iCast覆膜支架 l产品代码和序列号详见网站: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=193181。 l分销日期:2018年12月31日至2022年3月31日 l在美国召回的数量:68812 l召回发起日期:2022年3月3日 产品用途:iCast覆膜支架系统是一种球囊可扩张支架,可以使用导管输送以保持打开并支撑体内结构壁。在美国,该产品目前用作保持气道畅通的支架(气管支气管支架)。 召回原因:由于收到越来越多的客户投诉,当从人体上移除输送系统时,球囊或者导管縠与输送系统分离,Atrium Medical公司正在召回iCast覆膜支架。当支架系统在适应症外使用时,这个问题似乎最常发生,例如用于治疗血管疾病。 如果设备从患者血管中取出之前发生分离并且球囊没有完全放气,则可能导致手术时间比预期的要长,使患者暴露于额外的麻醉/成像造影剂中。在移除输送导管后,分离的部分也可能留在体内,导致血管闭塞,后续可能出现截肢、栓塞、器官功能丧失、器官梗塞和组织梗塞等不良后果。 目前,已收到75起投诉,其中9例伤害报告,尚无与此设备问题相关的死亡事件。 受影响人群: l使用iCast覆膜支架系统治疗患者血管疾病的医务人员,尤其是该产品在适应症以外用途使用的。 l使用iCast覆膜支架系统进行基于导管的血管手术的患者。 采取措施: 2022年3月3日,Atrium Medical公司向所有客户发送了一份紧急医疗器械更正通知,建议客户在使用此设备之前阅读以下放气和移除说明: l将充气装置上的真空拉至最大体积来使球囊放气,并留出足够的时间进行完全放气。放气时间可能因球囊尺寸、导管长度和使用的充气介质而异。对于更大的设备和更高的对比浓度,放气可能需要更长的时间。 l在尝试移除输送系统之前,通过荧光透视验证球囊是否完全放气。 l如果遇到阻力,不要强行移除输送系统。强制移除可能会导致对输送系统的损坏,包括球囊或者导管毂与输送导管的分离。 l如果无法使球囊完全放气或者遇到阻力,则将输送系统和导管鞘作为一个整体移除。建议在手术完成之前导丝一直穿过病灶。在保持导丝位置和充气装置负压的同时,缓慢地移除输送导管。 (美国FDA网站) 美国FDA发布关于Medtronic公司因泵焊接缺陷召回HVAD泵植入组件的警示信息 发布日期:2022年6月10日 召回级别:I级,是最严重的召回类型,使用这些器械可能造成严重伤害或者死亡。 召回产品: l产品名称:HVAD泵植入组件 l型号:1101、1103、1104、1104JP、MCS1705PU l分销日期:2006年10月11日至2021年6月3日 l在美国召回的设备:1614台 l公司发起日期:2022年4月11日 产品用途:HeartWare心室辅助装置(HVAD)系统用于帮助心脏继续向身体其他部位泵血。HVAD系统被用作有死于终末期左心室心力衰竭风险的患者心脏移植的桥梁,用于心脏组织恢复,或作为未计划心脏移植的患者的目标治疗(DT)。
召回原因:由于泵的焊接缺陷,Medtronic公司正在召回HVAD泵植入组件。在对退回给Medtronic的外植泵进行检查后,分析显示水分已经进入泵的中心柱,导致内部磁铁腐蚀和退磁,这可能导致泵旋转不正确。使用受影响装置的患者可能会出现类似于泵血栓形成的症状和体征。 如果出现这种问题,可能会导致泵故障、死亡或严重伤害(例如:伴有严重器官功能障碍的休克、中风),或者要求患者接受大手术来更换泵。 Medtronic公司已经收到了3起关于该设备问题的投诉,包括1起死亡和2起伤害事件。 受影响人群: l使用Heartware HVAD系统的医护人员 l使用Heartware HVAD系统进行手术的患者 采取措施:2022年4月11日,Medtronic公司发送了一封紧急医疗器械纠正函,以筛选出拥有未使用的HVAD泵植入套件的医生和医疗保健服务提供者,要求退回产品。 2022年4月26日,Medtronic公司向所有医生和医疗保健服务提供者发送了一封紧急医疗器械纠正函,通知他们这一潜在的泵焊接缺陷。 这封信要求客户: l当患者出现信函中列出的体征和症状时,将.csv日志文件全部上传并提交给Medtronic公司。 l考虑出现信中所列任何体征和症状的患者是否可能由泵血栓引起,并进行相应治疗。考虑到患者的临床情况和手术风险,医生应该根据具体情况决定是否移除或更换泵。 l将这封信分享给其组织内所有需要了解这一点的人,或转移了潜在受影响患者的任何组织。 l填写随附的客户确认表,并通过电子邮件发送至RS.CFQFCA@medtronic.com。 (美国FDA网站)
| |||
|