|
|
| 当前位置: 首页>首页>消费提示>医疗器械 |
| 医疗器械警戒快讯 2021年第3期(总第169期) | |||
| 分享到: | |||
| 发布日期:2021-03-18 | |||
|
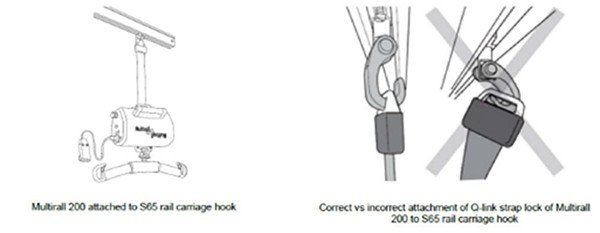
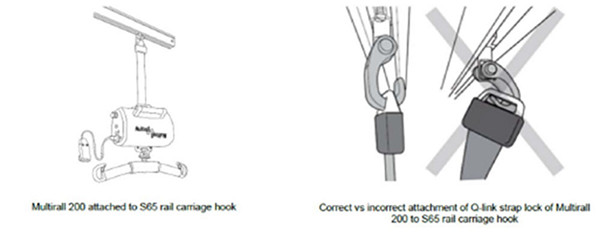
医疗器械警戒快讯 2021年第3期 (总第169期) 内容提要 ● 美国FDA发布关于美敦力公司因停止后重启延迟或失败风险召回HVAD泵植入套件的警示信息 ● 美国FDA发布关于美敦力公司因支架断裂风险召回Valiant Navion胸腔支架系统的警示信息 ● 美国FDA发布关于Hillrom公司因Q型连接带锁(也称为第一代Q型带锁)连接到S65吊钩失效风险召回Liko Multirall 200高架升降机的警示信息 ● 美国FDA发布关于Acist Medical Systems公司因产品损坏风险召回ACIST Kodama血管内超声导管的警示信息 ● 加拿大Health Canada发布Cardinal Health公司因空气进入风险召回肠道营养泵套件的警示信息 ● 美国FDA发布不正确使用热成像系统的安全性通报 ● 澳大利亚TGA发布关于BD公司因产品无菌性风险召回Alaris泵(GP、VP、CC、SE)用输液器及非专用输液器和配件的警示信息 ● 澳大利亚TGA发布关于Bard Australia公司因深度调节旋钮使用过程中与设备分离风险而召回一次性穿刺活检针的警示信息 国家药品不良反应监测中心 国家药品监督管理局药品评价中心 http://www.cdr-adr.org.cn 美国FDA发布关于美敦力公司因停止后重启延迟或失败风险召回HVAD泵植入套件的警示信息 发布日期:2021年3月1日 召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。 召回产品:HeartWare 心室辅助装置系统泵植入套件 产品型号: Medtronic HVAD泵植入套件,标示为:PUMP1103、PUMP1104、PUMP1104JP 产品用途:HeartWare心室辅助装置(HVAD)泵植入套件是HeartWare HVAD系统的一部分,用于帮助心脏继续向身体其他部位泵血。HVAD系统被用作终末期左心室心力衰竭死亡风险患者心脏移植的桥梁,用于心脏组织恢复,或作为未计划心脏移植患者的永久替代治疗(DT)。 召回原因:Medtronic正在召回HVAD泵植入套件,因为该设备可能无法初始启动、重新启动,或在泵停止后延迟重新启动。这些延迟、无法启动或重新启动发生在植入前测试、植入期间或各种植入后情况。如果设备延迟、无法启动或重新启动,这可能会导致严重的患者伤害,包括心脏病发作、心力衰竭恶化、需要额外的程序和住院治疗或死亡。 共有29起关于该设备问题的投诉,其中包括19起重伤和8例有生命危险事件但患者未受到长期影响。据报道有两人死亡。 召回措施:2020年12月18日,美敦力向所有受影响的客户发送了一封紧急医疗器械通信函;2020年12月23日,美敦力向之前购买过美敦力HVAD泵的所有账户发送了一封紧急医疗器械通信函。通知要求客户: 根据当前使用说明(IFU),建议医疗服务人员和工作人员注意以下几点,以避免不必要的停泵: 1不要从控制器上断开传动系; 2不要同时从控制器上断开两个电源(电池和交流或直流适配器);一个外部电源应始终与控制器保持连接; 3除非高优先级报警条件或VAD团队成员明确指示,否则不要更换控制器; 4加强对[控制器故障]警报和[电气故障]警报的正确响应。这些是与泵立即停止无关的中等优先级警报。这些警报将在控制器显示屏上显示单词[Call],通知患者呼叫他们的临床医生; 5加强电源和控制器端口数据电缆的连接; 在任何控制器更换之前,通知植入这些已识别泵之一的患者联系他们的心室辅助装置协调员,并在临床环境中协调执行控制器更换; 决定是否有必要为植入这些已识别泵的患者更换控制器,并考虑以下事项: 控制器更换应在临床医生的监督下,在一个可控的环境中进行,并能立即使患者获得血液动力学支持。无法重新启动可能是致命的; 泵停止后,高优先级[VAD Stopped]警报将在控制器显示屏上显示[Change Controller]或[Connect Driveline]。重新建立电源和传动系连接后,如果泵未重新启动: 考虑当前控制器的电源循环或考虑控制器更换。这将允许重新启动算法复位并重新启动。控制器最多自动尝试重新启动泵30次;[VAD Stopped]警报在五(5)次尝试后开始。 如果泵仍未重新启动,则继续进行临时血液动力学支持和泵更换。 在内部控制器电池达到使用寿命并触发[控制器故障]警报(如果患者的控制器使用超过两(2)年)之前安排控制器更换; 尽管[控制器故障]报警是与停泵无关的中等优先级报警,但主动安排控制器更换有助于避免患者在临床之外更换控制器,从而对报警作出反应。根据IFU,患者应在收到中等优先级警报时致电临床医生。 查看信件中的序列号,确认患者是否仍在接受帮助范围内; 与所有需要了解的人分享这封信; 填写医生确认表(随信附上)并通过电子邮件发送至RS.CFQFCA@medtronic.com。 (美国FDA网站) 美国FDA发布关于美敦力公司因支架断裂风险召回Valiant Navion胸腔支架系统的警示信息 发布日期:2021年3月10日 召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。 召回产品:Valiant Navion胸腔支架系统,具体召回产品编号见FDA网站。 产品用途:该产品被用于降主动脉病变的血管内修复。 召回原因:出现IIIb型内漏、支架断裂和支架环扩大。 召回措施:2021年2月12日前后,美敦力公司向全球客户(风险管理者和医生)发出了口头通知,以立即停止植入并隔离任何尚未使用的受影响产品。 (美国FDA网站) 美国FDA发布关于Hillrom公司因Q型连接带锁(也称为第一代Q型带锁)连接到S65吊钩失效风险召回Liko Multirall 200高架升降机的警示信息 发布日期:2021年2月22日 召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。 召回产品:Hillrom的Liko Multirall 200高架升降机 产品型号、产品代码、目录或批号: Liko Multirall 200(产品编号3130001) 通用吊带450 R2R(产品编号3156095) 通用吊带350 R2R(产品编号3156094) 带双钩的D45吊厢(产品编号3136100) 延伸带300-400毫米(产品编号3136226) 延伸带400-600毫米(产品编号3136227) 延伸带600-1000毫米(产品编号3136228) 延伸带1000-1400毫米(产品编号3136229) 制造日期:2000年12月至2020年10月 销售日期:2000年12月17日至2020年10月1日 美国召回设备:1160 公司发起日期:2020年12月18日 器械用途: Liko Multirall 200高架升降机是一种通用升降机,用于将患者从一个房间移动到另一个房间。这种升降机是Multirall 200高架升降机系统的一部分,该系统有一个高架升降机电机、S65轨道车吊钩和一个Q型连接带。该系统用于疗养院、康复医院等医疗保健机构。 软体高压氧舱可能会使病人或产品附近的人受到严重伤害,包括死亡。风险包括火灾、通过使用者之间的交叉污染传播传染病、耳朵、眼睛、鼻窦、肺和牙齿的损害,以及血糖水平的变化。当该装置与氧气浓缩设备结合使用时,火灾风险显著增加。
召回原因: Hillrom正在召回Liko Multirall 200高架升降机,因为客户报告说,Q型连接锁没有如预期一般连接到S65车架钩上。如果带锁没有连接,电机或患者就可能会跌落。使用受影响的产品可能会导致严重伤害和死亡等不良事件。 已经有34起关于该器械问题的投诉和22起严重伤害或死亡不良事件报告,其中有两例死亡。 影响人群 使用受影响的Hillrom器械的医务人员 使用受影响的Hillrom器械患者或患者身边的人员 召回措施: 2020年12月18日,Hillrom向所有受影响的客户发送了一封医疗器械紧急纠正函,为医疗服务提供商提供以下说明: 检查机构中的每一个Multirall装置,看它属于哪一类(A,B或者C)。 类别A:Multirall用于房间与房间相连的附件,如通用吊带350 R2R、通用吊带450 R2R或带双钩的房间间导轨架D45。 类别B:Multirall与一个延长带相结合。 类别C:Multirall不与A类或B类中的任何附件结合。 填写随信提供的反馈表,并在一个月内将其发送给Hillrom(MOD1322@hillrom.com)或Hillrom经销商。 酌情将此通知分享给其他组织。为确保有效性,在一定时间内持续关注本通知及相关信息。 一旦发现上述器械并收到反馈表,Hillrom或其官方经销商将联系客户,用第二代Q型连接带更换Q型连接带。 (美国FDA网站) 美国FDA发布关于Acist Medical Systems公司因产品损坏风险召回ACIST Kodama血管内超声导管的警示信息 发布日期:2021年3月4日 召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。 召回产品:Acist Medical Systems公司生产的ACIST Kodama血管内超声导管 产品型号:017788、018125(仅限日本);批号:00233370(100件)、00233371(90件)、00233372(100件)、00233373(100件)、00233374(100件)、00233380(100件)、00233384(60件)、00233385(100件)、00233393(100件)、00233394(100件)、00237604(35件)、00237613(100件)、03012517(100件) 产品用途:Kodama血管内超声导管是ACIST HDi系统的一个组成部分。ACIST HDi系统用于冠状动脉和周围血管病变的超声检查,筛选开展血管内介入治疗的患者。ACIST-Kodama血管内超声导管适用于ACIST-HDi系统。 召回原因:Acist Medical Systems公司通过生产线的测试发现,部分Kodama血管内超声导管存在一块损坏的o形圈。进一步的测试表明,损坏的o型环碎片(>200微米)有可能被冲出导管。ACIST Medical Systems公司正在确认故障模式的来源,以确保Kodama血管内超声 导管的质量和可靠性。目前公司没有与此事件相关的现场报告,也没有任何关于患者受伤或不良健康后果的证据或报告。 召回措施:ACIST Medical Systems公司向客户发送了“紧急:医疗器械召回”信函和回复表。要求客户执行以下操作:①查看Kodama 导管清单;②在响应表中记录每批的数量 ;③从库存中删除受影响的批次;④使用随附的预付退回标签退回受影响的产品,包括随产品提供的回复表副本。 (美国FDA网站) 加拿大Health Canada发布Cardinal Health公司因空气进入风险召回肠道营养泵套件的警示信息 发布日期:2021年3月1日 召回级别:II级 召回产品:Kangaroo 肠道营养泵套件 召回原因:产品在安装期间管道中可能出现空气。 召回措施:Cardinal Health公司正在发起一项医疗器械产品警告通信,以告知客户此通信的目的是将此情况告知客户,并在获得改进后的产品之前为客户提供解决问题的步骤。 (加拿大Health Canada网站) 美国FDA发布不正确使用热成像系统的安全性通报 发布日期:2021年3月4日 产品用途: 用于测量人体皮肤表面温度的热成像系统按照联邦食品、药品和化妆品法案第201(h)节的规定进行管理。这些系统检测从人体皮肤发出的红外光,然后将这些信息转换成皮肤温度读数。然后,可以使用皮肤温度来估计对应的身体部位(例如,嘴和耳朵)的温度。 通报内容: 美国食品和药物管理局(FDA)警告消费者、医疗机构和其他使用热成像系统测量人体温度的用户,不当使用这些医疗器械可能会造成温度读数不准确。这些设备也被称为远程热成像系统、红外热像仪、热感摄像机和“发热照相机”。据我们所知,热成像系统目前正被用于公共场所(如机场、工作场所、零售商店、学校)的初步温度评估和发热检测,同时,这种系统可以与包括社交距离、戴口罩和勤洗手在内的其他措施相结合,成为新冠肺炎风险管理总体措施的一部分。 发热或体温升高只是新冠肺炎感染的一种可能症状。虽然热成像系统在正确使用的情况下通常能准确检测到体温升高,但它们不能检测到任何其他感染症状。据了解,并非所有新冠肺炎阳性个体都会出现体温升高,因此体温不应成为疾病评估中的唯一体征或症状。因此,包括使用热成像系统在内的温度测量,不应作为诊断或排除新冠肺炎疾病的单一或主要依据。确定某人是否患有新冠肺炎必须进行诊断试验。 热成像系统的不当使用可能导致体温测量不准确,从而带来严重的公共卫生风险。这些风险可能包括但不限于,当体温升高检测为正常体温,或将正常体温检测为体温升高。当热成像系统同时扫描多个个体时,这些风险更有可能出现。为了帮助减轻这些风险,FDA向消费者、医疗机构和其他用户提供了关于正确使用这些系统的重要建议。 针对热成像系统消费者、医疗机构和其他用户的建议 ● 一次只测量一个人的体温。迄今为止,还没有证据显示这些系统在用于同时测量多人体温时是有效或准确的。因此,它们不应用于“批量体温筛查”。 ● 更多关于热成像系统(红外热成像系统/热成像摄像机)的使用注意事项和最佳实践信息,包括但不限于: 热成像系统的准确性取决于仔细的设置和操作。 这些设备测量的是皮肤表面温度,因此在环境变化时需要一段时间使被测者的皮肤适应,例如,从室外进入建筑物时。其中一些环境因素包括气温、湿度、风和阳光。 使用热成像系统的环境温度应该在68-76华氏度(20-24摄氏度)之间。这个范围之外的环境温度可以改变皮肤温度,导致测量结果可能不能反映人体内部体温。此外,房间不应该有风(空气流动),因为它会改变皮肤温度。 被测量的人不应该戴帽子或眼镜,同时避免头发遮挡脸部。FDA认为在新冠肺炎紧急公共卫生事件期间使用热成像系统时正确佩戴口罩的好处超过了潜在测量不准确的风险。 热成像系统不应在强光源附近使用,如阳光或明亮的灯光。此外,系统不应面对任何能反射光线的东西,如窗户或其他反光表面。 高温读数应该用数字温度计进行确认,例如测量舌下(口腔)、手臂下(腋窝)或耳部(鼓膜)的读数。 向医务人员咨询体温升高的意义。 热成像系统不应用于诊断新冠肺炎,但当作为风险管理总体措施一部分时,其使用可被视为初始温度评估的方法。 热成像系统使用不当的风险 如上所述,热成像系统在用于同时测量多人温度时的准确性未经确认,因此不应用于“大规模温度筛查”。这些器械的准确性取决于仔细的设置和操作以及被评估人的适当准备。这些器械的不当使用可能会导致皮肤温度读数不准确。与不准确的温度测量相关的风险可能包括漏掉皮肤温度升高的被测者,或者错误地将一个体温正常的被测者评估为皮肤温度升高。 FDA的相关行动 ● 2020年4月,FDA发布了《新冠肺炎突发公共卫生事件期间远程医疗系统的执法政策》,帮助扩大热成像系统的可用性,缓解紧急公共卫生情况下温度计的短缺。该指南规定了一项适用于新冠肺炎紧急公共卫生情况期间用于医疗目的的所有热成像系统的强制政策,并提供了关于此类系统的性能和标签的建议。 ● FDA了解到有一些公司销售未经批准、备案或授权的用于测量人体温度包括同时测量多个人的温度的热成像器械。FDA已经发布了几个相关的警告信,必要时可能采取其他行动。 ● FDA的立场是,警告信仅针对具有监管意义的违规行为发出。相关警告信发布在专门的警告信网页。新冠肺炎相关的警告信发布在FDA新冠肺炎相关产品欺诈的页面。 ● FDA将继续监测这一问题,如果有重要信息更新,将随时向公众通报。 报告器械问题 如果想报告热成像系统的问题,FDA鼓励填写MedWatch自愿报告表进行报告。 符合FDA报告要求的机构所雇佣的卫生保健人员应遵循其机构制定的报告程序。 额外资源 ● 关于新冠肺炎的更多信息,包括症状、检测或何时应获取医疗帮助的信息,请访问疾病预防和控制中心新冠肺炎页面。 ● 若要获得更多关于热成像系统的获益、限制和正确使用的信息,请访问FDA热成像系统页面。 (美国FDA网站) 澳大利亚TGA发布关于BD公司因产品无菌性风险召回Alaris泵(GP、VP、CC、SE)用输液器及非专用输液器和配件的警示信息 发布日期:2021年3月10日 召回级别:Ⅰ 级 召回编号:RC-2021-RN-00707-1 召回产品:Alaris泵(GP、VP、CC、SE)用输液器及非专用输液器和配件 产品注册号:189736-BD-导管延长装置,静脉注射 125916-BD-输液管理器,输液泵 177500-BD-输血器 246696-BD-负压无针阀接头 299777-BD-基本静脉给药装置 召回原因:BD被第三方灭菌服务提供商告知,他们故意伪造了与BD产品加工相关的灭菌过程记录。 BD立即开展了调查,但无法保证上述产品的无菌性,因此决定将这些产品从市场撤回。 召回行动: 请客户和用户按照告知信函中的说明立即封存和处理受影响的产品,BD会对受影响的产品进行替换。 (澳大利亚TGA网站) 澳大利亚TGA发布关于Bard Australia公司因深度调节旋钮使用过程中与设备分离风险而召回一次性穿刺活检针的警示信息 发布日期:2021年2月25日 召回级别:Ⅱ级 召回编号:RC-2021-RN-00597-1 召回产品:一次性穿刺活检针 产品注册号:302839 召回原因:Bard Australia公司通过对产品投诉的审查,发现投诉报告中,穿透深度开关(深度调节旋钮)在使用过程中与设备完全分离的问题趋势增加。深度调节旋钮的作用是能够选择18mm或25mm的理想穿透深度。 这个问题可能导致手术时间延长,穿透不足导致无法获取样本,或穿透过度导致邻近组织损伤。目前,澳大利亚已收到25份与该问题有关的投诉报告,未收到不良事件的报告。 召回行动:建议客户与召回通知中的相关人员进行沟通联系,并立即检查库存中具体的产品代码和批号。完成确认表格邮件发送至ANZ_Quality@bd.com,同时正确处置受影响的产品。 收到确认表后,BD代表将联系客户,为受影响的库存安排信贷。
| |||
|